

If genes could talk
Genomic sleuthing paints pictures of both past and present
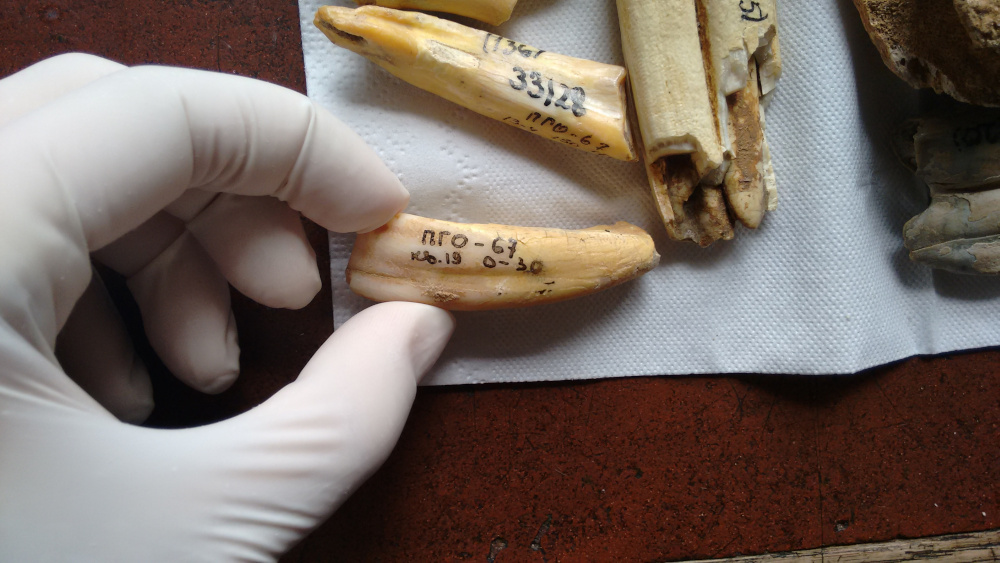
More than a million years ago, snow and ice buried a mammoth’s decaying body in Siberia. Over time, most of the “Krestovka mammoth” vanished; first its soft tissues and later even its bones became food for bacteria and bugs. Some teeth remained, however, and a Russian paleontologist unearthed a few in 1970. For decades, they sat in museum collections as curiosities, too old to carbon date. Scientists presumed any lingering organic material long gone.
However, in research published in February 2021 in the scientific journal Nature, an international team of scientists including Beth Shapiro, professor of ecology and evolutionary biology, turned to modern genomics to reveal more about this ancient mammoth and its relatives. Drilling a miniscule amount of material—about a pinch of salt’s worth—out of one molar, the team was able to isolate tattered bits of DNA from the sample. Their sequencing of this and other mammoth DNA showed that the Krestovka specimen represented a previously unknown genetic lineage of mammoths, upending previous hypotheses about mammoth evolution. Based on its sequenced genes and how much they varied from those of more modern mammoths, the team estimated that the animal lived about 1.2 million years ago. Before this effort, no one had ever sequenced genes more than a million years old.
Until recently, such a genomic dive into the deep past was considered impossible. But gene sequencing continues to get better, faster, and cheaper, allowing scientists, like Shapiro and others at UCSC, to develop new ways to piece together and analyze genes from even the most fragmented DNA specimens. The advances are letting researchers solve long-standing mysteries about evolution, migration, and diversity—in both animals and humans. They’re also a boon for understanding and diagnosing genetic diseases and helping criminal investigators solve cold cases. “Even a single genome can tell us all kinds of things about the broader population it belongs to,” said Shapiro. “It really can paint a picture of the past.”
Dog-chewed

{kind=link}
When people swab the inside of their cheeks with test kits like those from 23andMe and ancestry.com, they collect—among other things—strands of genetic material, each containing hundreds of millions of relatively intact nucleotide base pairs, the building blocks of DNA. Sequencing such DNA samples is straightforward. But immediately after an organism dies, the natural world around them starts breaking down their components, including their DNA. Further muddling the genomic picture, microbes and other decaying plants and animals seep into the heap, adding their DNA remnants.
If sequencing DNA from a living person is like putting together a thousand-piece puzzle per the picture on the box cover, then sequencing ancient DNA is like solving such a puzzle after your dog chews up each puzzle piece, someone mixes those pieces with dog-chewed pieces from several other puzzles, and you lose the box. “To sequence ancient genomes, we need to take dirty, crumbled up DNA and transform it into molecules that can be sequenced,” said Shapiro.
Shapiro and her husband Richard Edward Green, associate professor of biomedical engineering, now routinely perform this difficult task in the UCSC Paleogenomics Laboratory they run together. The work involves tedious washing, isolating, and handling to get rid of contaminants, gene amplification to make millions of copies of tiny bits of DNA, and powerful computer programs that assemble the short sequences into more complete gene readouts. “We need specialized facilities where we don’t touch our samples,” said Shapiro. “We don’t even breathe on them—if our DNA gets into this ancient DNA confetti, it makes our experiments much harder.”
Assessing admixing

{kind=link}
Work at the Paleogenomics Laboratory in the last few years has sequenced not only the genomes of mammoths but also those of ancient bears, dogs, bison, horses, and Neanderthals. A key question tying many of the projects together is what genetically defines a species. “As humans, we have this proclivity to put things into boxes—a polar bear is a polar bear, and a brown bear is a brown bear,” said Shapiro. But this view is too simple. “Speciation typically doesn’t happen overnight.”
In the genomes of nearly all the ancient animals Shapiro and others have studied, the results indicate that those from different lineages often interbred even after beginning to separate into distinct species, a phenomenon called admixing. The result? Brown bears have snippets of polar bear DNA, bison have cattle in their genomes, and modern humans share a surprisingly large amount of DNA with Neanderthals.

{kind=link}
Cataloguing these genetic admixtures can help scientists understand how ancient populations arose and declined. But it also can prove valuable in conservation efforts to help save struggling species from extinction. In some cases, preventing admixing might be critical to ensuring the survival of a rare species—if two species interbreed too frequently, the unique traits of one may completely fade away. More often, though, endangered populations suffer from interbreeding between close relatives, which can harm a population by increasing the prevalence—and impact—of detrimental genes.
To make at-risk species healthier and more likely to survive long term, conservationists sometimes encourage the breeding of isolated populations with closely related animals from the outside. Scientists successfully boosted Florida’s dwindling panther population in the 1990s, for instance, by cross-breeding the Florida animals with Texas cougars. Having genetic information on each species helps conservationists choose which populations—or even individual animals—to breed with each other, which can be critical to the success of such efforts. “The past is a completed evolutionary experiment,” said Shapiro. “But when we try to devise strategies to protect species, we're making a lot of guesses. By looking into the past, we can better understand what has already worked.”
Seeing differences
The genomic science advances enabling scientists to study ancient DNA and help endangered animals also have vast promise to improve human health, said Benedict Paten, associate professor of biomolecular engineering and associate director of the UCSC Genomics Institute. Some of that promise is clearcut. For example, as reported in February 2022 in the New England Journal of Medicine, Paten and collaborators showed that rapid sequencing of the entire genomes of critically ill patients uncovered genetic variations that impacted the treatment of five of the dozen subjects enrolled in the study. But other implications are less straightforward. Understanding human genetic evolution, Paten said, can shed light on how today’s diversity arose and how it impacts human health, which could help address the increasingly acknowledged—yet quite elusive—problem of systemic racial bias in medicine.
Paten chairs the Genome Alignment and Annotation Committee of the Vertebrate Genomes Project, a group of more than 150 scientists, each with their own funding, aiming to collaboratively sequence at least one genome from 66,000 vertebrate species, including mammals, birds, reptiles, amphibians, and fish. Like Shapiro and Green, Paten is not only interested in the stand-alone genomes of single species, but how these genomes compare and have evolved and diverged over time. “On their own, genomes can be incredibly interesting but they’re also snapshots in time in a long evolutionary history,” Paten said. Comparing the genomes of different species can reveal new genes and proteins that could lead to the discovery and development of a broad range of innovative items, from medicines to adhesives or even electronics, said Paten. “Every species contains unique genes that can be powerful in terms of understanding biology and giving us new tools.”
In addition, understanding how and why one gene has evolved to vary between species while another has remained stubbornly stagnant can tell Paten and other researchers much about the importance—and flexibility—of that gene. Paten gauges this flexibility by comparing variation between modern species, while Shapiro and researchers in the Paleogenomics Lab often try to assess the same thing by analyzing the genomes of ancient species. In July 2021, for instance, the journal Science Advances published Shapiro and Green’s research comparing modern human and Neanderthal genomes to see which parts of the modern human genome were never—in any individuals—derived from Neanderthal ancestry. Less than 7% of the modern human genome, they discovered, is unique and never of Neanderthal origin. “Some traits move between species and become advantageous, but others don’t, because they can’t. They’re not compatible,” said Shapiro, adding that knowledge of a gene’s natural variation could eventually prove helpful to bioengineers attempting to repair or improve its function.
Pushing envelopes
If genomes could talk, each human’s would sound different. Diversity in ancestry, race, disease risk, metabolism, and a plethora of other features, from personality to appearance, are all captured in our DNA. But for the past nearly 20 years since the milestone completion of the Human Genome Project in 2003, researchers have used a single reference genome—based almost entirely on the genome of a single individual from Buffalo, New York—as the template for all other human genomes. “That’s a problem,” said Karen Miga, Paten’s colleague, and an assistant professor of biomedical engineering. “The reference genome gives us only one snapshot of a human genome when we all know that our genomes differ, sometimes in very large ways.”
When researchers analyze a person’s genome, they use that reference genome as a point of comparison. Places where an individual’s genes vary greatly from the reference may indicate causes of disease, for instance. But for some populations, the current reference genome may not be a fair place to start—it may not capture the norm at all.
Like Shapiro, Green, and Paten, Miga is pushing the envelope on how to obtain the most detailed information from genomes and capture the differences between them. Miga co-chaired the Telomere-to-Telomere Consortium, an effort—just recently completed and reported in six papers published in the March 31, 2022 issue of Science—to fill in the gaps that remained in the original reference genome. She also co-chairs working groups within the broader Human Pangenome Project, which aims to create a more global reference genome that better represents human genomic diversity. In all this work, Miga uses multiple new technologies, including nanopore sequencing, that let researchers piece together much larger chunks of the genome at a time—akin to completing a puzzle with a hundred pieces instead of one with a thousand pieces. This makes it easier to figure out the many repetitive stretches of DNA within people’s genomes—a challenge that’s a bit like assembling a large blue sky of a puzzle.
The rapidly improving technology, Miga said, doesn’t just benefit the Pangenome Project, but gene sequencing for all other purposes. This broad applicability rings true for all the researchers. The same breakthroughs that allow them to determine how horses or wolves evolved can also help them better diagnose patients with rare diseases, identify criminals, and engineer improved food crops. “A lot of basic research has a very long tail,” said Paten. “Genomics often asks some fairly technical questions about how we can assemble the whole genome and all its variations, but the progress we make on those questions serves us well in many, many other areas.”